Inteligencia artificial revoluciona la biomedicina
Un sistema que emplea esta tecnología para predecir estructuras 3D de proteínas impulsa el desarrollo de nuevos fármacos.
El Premio Lasker de Investigación Médica Básica 2023 realza el valor de un sistema de inteligencia artificial que predice la estructura tridimensional de las proteínas a partir de su secuencia unidimensional de aminoácidos. Esta propuesta abre un camino hacia grandes descubrimientos en las ciencias biomédicas en un menor tiempo.
Las proteínas desempeñan un papel fundamental en las enfermedades, por ejemplo, se pliegan incorrectamente y se agregan en el Alzheimer, se desregulan en los cánceres, son disfuncionales en los errores congénitos del metabolismo y se transportan al compartimento equivocado en la fibrosis quística. Estos son sólo algunos de los diversos mecanismos. Los modelos estructurales detallados de proteínas aceleran el descubrimiento de fármacos al proporcionar estructuras atómicas, que impulsan el diseño o la selección de moléculas de alta afinidad.
Las estructuras de proteínas se determinan experimentalmente mediante cristalografía de rayos X, resonancia magnética nuclear, y microscopía crioelectrónica. Estos métodos son costosos y requieren mucho tiempo. En consecuencia, la base de datos de estructuras 3D de proteínas tiene aproximadamente 200.000 estructuras, mientras que la tecnología de secuenciación de ADN ha producido más de 8 millones de secuencias de proteínas. En la década de 1960, Christian B. Anfinsen y colaboradores demostraron que la secuencia de aminoácidos puede plegarse espontánea y reproduciblemente en la conformación funcional 3D (figura 1A). Las “chaperonas” moleculares pueden acelerar y facilitar este proceso, pero estas observaciones crearon un gran desafío para la biología molecular: predecir la estructura 3D de una proteína a partir de su secuencia de aminoácidos. Este desafío se volvió más apremiante a medida que la capacidad para obtener secuencias 1D se disparó con el éxito del Proyecto Genoma Humano.
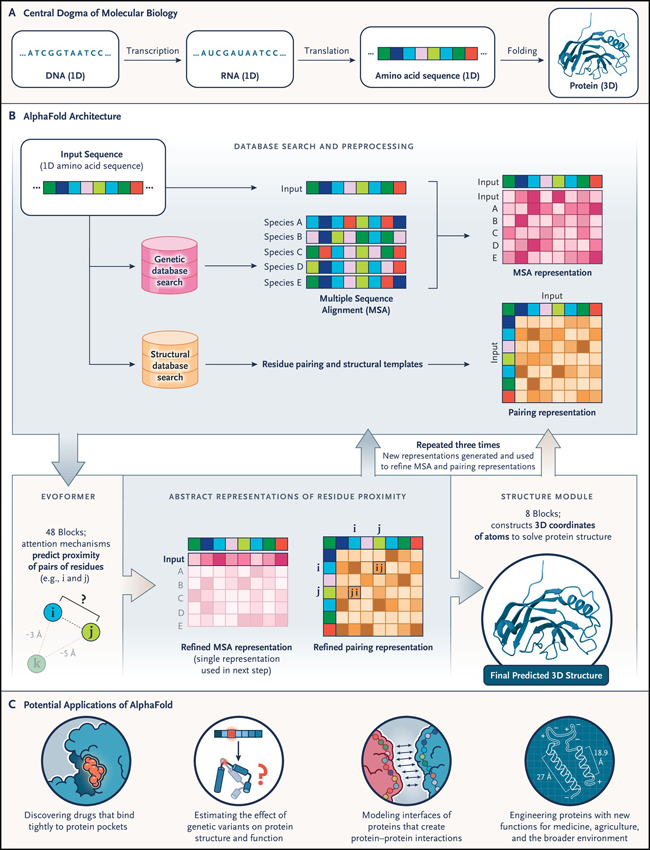
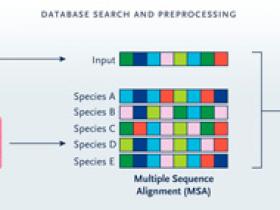
Figura 1: estructura de las proteínas con AlphaFold.
La predicción de la estructura proteica es compleja por varias razones. En primer lugar, existe un gran espacio de búsqueda: todas las posiciones 3D posibles para cada átomo de cada aminoácido. En segundo lugar, las proteínas maximizan químicamente las interacciones complementarias para configurar sus átomos de manera eficiente. Encontrar posiciones en las que casi todos los donantes estén cerca de un aceptor puede ser exponencialmente difícil. Por último, hay ejemplos limitados de modelamiento a partir de métodos experimentales y, por lo tanto, se debe aprovechar la información evolutiva de proteínas relacionadas, para comprender posibles interacciones 3D entre aminoácidos sobre la base de la secuencia 1D.
En un enfoque para predecir la estructura de proteínas, en base a la física, se modela las interacciones de los átomos mientras se busca configuraciones óptimas. Karplus, Levitt y Warshel recibieron el Premio Nobel de Química en 2013 por sus contribuciones en la simulación computacional de proteínas. Sin embargo, los métodos basados en la física son costosos y requieren aproximaciones, por lo tanto, no presagian estructuras 3D precisas a escala. En otra perspectiva, "basada en el conocimiento", se utilizan bases de datos de estructuras y secuencias conocidas para perfeccionar modelos de proteínas con el uso de inteligencia artificial y machine learning (IA-ML). Hassabis y Jumper incluyeron elementos tanto de física como de IA-ML, proporcionando este último mejor aproximación y mayor rendimiento. Combinaron grandes repositorios de datos públicos con recursos computacionales a nivel industrial para construir “AlphaFold”.
¿Cómo sabemos que "resolvieron" el problema de la predicción de estructuras? Existe una comisión que evalúa esta predicción y ha documentado saltos periódicos en el rendimiento en base a las innovaciones metodológicas. Las predicciones de AlphaFold presentadas en 2020, mostraron un salto en el rendimiento tan grande que los organizadores dieron por resuelto el problema de la predicción de estructuras 3D. La mayoría de las predicciones tenían una precisión similar a la de las determinaciones experimentales.
¿Cómo lograron tales resultados? Los investigadores realizaron una serie de innovaciones técnicas, incluido un proceso de predicción de extremo a extremo (1D a 3D) que es diferenciable, de modo que todos los parámetros de su modelo se pueden optimizar simultáneamente (figura 1B). Las ideas propuestas sentaron la base para una serie de innovaciones.
AlphaFold ya se está utilizando para impulsar la creación de nuevos medicamentos (figura 1C). Ahora se pueden modelar estructuras nunca vistas en busca de su función. Los diseñadores de proteínas lo están utilizando para perfeccionar sus diseños de proteínas genéticamente modificadas. Este sistema también genera estructuras iniciales que se perfeccionan con datos experimentales, permitiendo obtener modelos para grandes máquinas celulares. Tiene algunos elementos sin resolver (por ejemplo, el modelado de proteínas mutantes), sin embargo, proporciona un punto de partida para avanzar en estos procesos específicos.
En conclusión, este trabajo demuestra claramente cómo la IA-ML está transformando la ciencia. Esta tecnología puede construir hipótesis científicas complejas a partir de múltiples fuentes de datos, correlacionar información clave, y determinar la calidad de sus datos. IA-ML es básicamente hacer ciencia.
Fuente bibliográfica
A Holy Grail — The Prediction of Protein Structure
Russ B. Altman, M.D., Ph.D.
Departments of Bioengineering, Genetics, Medicine, and Biomedical Data Science, Stanford University, Stanford, CA.
DOI: 10.1056/NEJMcibr2307735