Implicaciones clínicas de la leptina en la obesidad
La obesidad infantil es una condición desafiante. Las causas secundarias se buscan, pero rara vez se encuentran, y solo algunas pueden abordarse mediante intervenciones terapéuticas exitosas.
Esta hormona proteica alguna vez fue descrita como un tratamiento destacado, pero rápidamente perdió popularidad cuando se observó resistencia. En una reciente publicación (N Engl J Med 2023;388: 2253 - 2261) se describen variantes etiológicas en el gen de la leptina (LEP) en dos niños con sobrepeso severo cuyas condiciones eran resistentes a la terapia con metreleptina (sustituto farmacológico de la leptina), evidenciando una exitosa estrategia de tratamiento, con considerables implicaciones para la obesidad.
La leptina es una hormona peptídica generada predominantemente en el tejido adiposo. El almacenamiento del exceso de triglicéridos conduce a la expansión de las gotitas de lípidos durante la alimentación; como resultado, los adipocitos secretan leptina a la circulación (figura 1). Después de cruzar la barrera hematoencefálica, inhibe las neuronas orexigénicas en el hipotálamo al unirse a la forma larga de su receptor presente en la superficie de estas neuronas. Por tanto, tiene el efecto de incrementar el gasto de energía y disminuir la ingesta de alimentos. Sin embargo, a medida que aumentan sus niveles en la circulación, la hormona se vuelve menos efectiva para inhibir las neuronas orexigénicas.
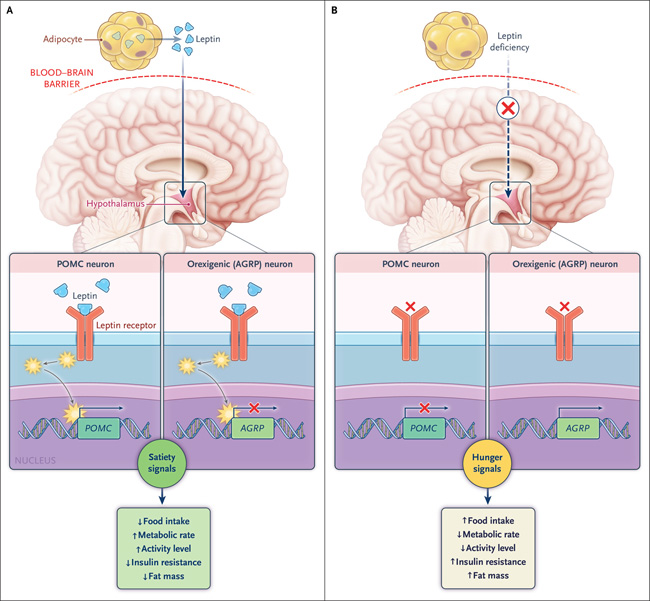
Figura 1: señalización, metabolismo y obesidad de la leptina.
Una cepa de ratones obesos, descrita por primera vez en 1950, permitió la comprensión de la obesidad adquirida genéticamente. En 1973, Doug Coleman descubrió que había una base genética para el fenotipo obeso de estos animales (llamados ratones “ob/ob”). Dos décadas después, se descubrió el gen causante: Lep, que codifica la leptina. En los seres humanos, su deficiencia congénita provoca un trastorno autosómico recesivo en el que las variantes genéticas de pérdida de función en LEP dan como resultado la ausencia total de leptina y, por tanto, una obesidad grave de aparición temprana.
Otra cepa de ratones mutantes, también descubierta hace más de 70 años, son modelos tanto la obesidad como la diabetes tipo 2. Estos tienen variantes inactivantes en el gen que codifica el receptor de leptina. Existen 38 variantes conocidas en el gen del receptor de leptina (LEPR) en humanos que causan obesidad severa de aparición temprana y diabetes tipo 2. En estas personas, las concentraciones de leptina circulante son muy altas; indicativo de resistencia a la leptina.
La resistencia adquirida a la leptina debido a la obesidad sin una causa genética conocida se caracteriza por niveles elevados de leptina sérica (figura 2). Los niveles persistentemente altos conducen a un antagonismo parcial y una bioactividad reducida. Un fenómeno similar ocurre en la mayoría de los individuos con obesidad que son tratados con metreleptina. Aquí, también, se cree que el antagonismo parcial entre la leptina endógena y la metreleptina causa resistencia a la metreleptina, aunque el mecanismo no está claro. Puede haber cambios postraduccionales en la molécula de leptina o alteración de su transporte a través de la barrera hematoencefálica. En teoría, la reducción de sus niveles (para reducir el antagonismo parcial) podría reducir la resistencia.
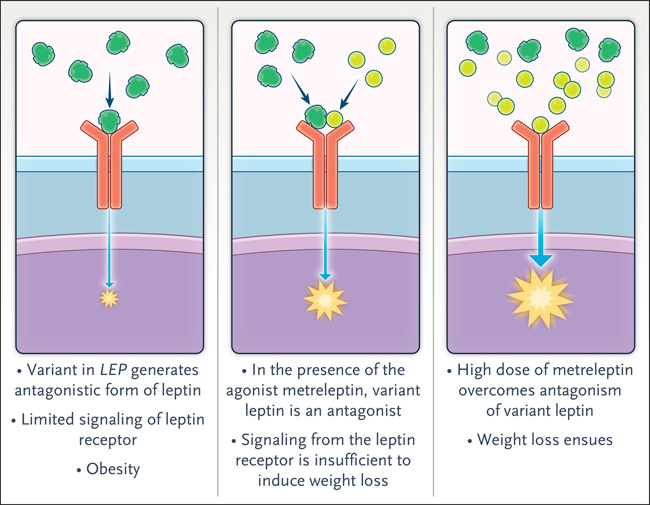
Figura 2: el agonista y el antagonista parcial.
¿Puede una molécula natural antagonizar una hormona endógena? Sí. En 1996, se comunicó el caso de un paciente con talla baja que presentaba una mutación puntual en el gen de la hormona del crecimiento. Esta resultó en una variante de la hormona del crecimiento que “superó” in vitro a la hormona del crecimiento no variante (codificada por la otra copia no mutada del gen de la hormona del crecimiento). Sin embargo, nunca se demostró de manera concluyente la interferencia con la señalización de la hormona del crecimiento, lo que llevó a dudar de la existencia de antagonistas competitivos naturales de una hormona endógena. La obesidad severa, la hiperfagia y los niveles elevados de leptina circulante en los niños descritos fueron causados por proteínas de leptina antagónicas que se unieron al receptor de leptina, pero desencadenaron una señalización marginal, si es que hubo alguna. En presencia de la leptina no variante, estas proteínas actuaron como antagonistas competitivos.
Para comprender el mecanismo molecular de las variantes LEP, vale la pena revisar algunos conceptos básicos en farmacología. El agonismo es la unión de un agente a un receptor que imita la actividad completa del compuesto endógeno, mientras que el agonismo parcial denota solo una parte de la actividad de señalización de ese compuesto (figura 2). El antagonismo competitivo ocurre cuando hay competencia por el mismo sitio de unión en el receptor que el agonista. Ninguna de las proteínas variantes de leptina inicia la señalización a través del receptor de leptina in vitro, aunque las afinidades de unión entre estas y el receptor de leptina son similares a las de la leptina nativa. Sin embargo, en los ensayos de unión competitiva, cada variante de leptina suprimió la señalización de la leptina nativa, por lo que cada una actuó como un antagonista competitivo de la leptina. En ausencia de leptina no variante (como era el caso de los pacientes, cada uno de los cuales portaba dos alelos variantes), cada variante tenía actividad agonista parcial.
Inicialmente, los pacientes fueron tratados sin éxito con dosis convencionales de metreleptina. Guiados por los resultados de sus experimentos bioquímicos, los investigadores aumentaron notablemente la dosis para superar los efectos del antagonista endógeno leptina. Además, los pacientes participaron en programas de ayuno y ejercicio para reducir la producción del antagonista endógeno leptina. Ambos perdieron mucho peso y se redujeron las dosis de metreleptina.
¿Qué nos dice estos resultados? Primero, la hormona variante endógena circulante puede antagonizar competitivamente a la hormona no variante (o su sustituto terapéutico, en este caso, metreleptina). En segundo lugar, se podrían considerar otras estrategias de tratamiento para la obesidad inducida por la dieta que involucren la leptina. Otros ensayos han analizado ratones con solo una copia intacta del gen de la leptina que recibían una dieta rica en grasas. En estos, los niveles de leptina no aumentaron en respuesta a la dieta, pero no se desarrollaron obesidad, hígado graso ni desregulación metabólica, lo que implica que los niveles de leptina comparativamente bajos pueden ser críticos para mantener la sensibilidad a la leptina. Es evidente que existe un estrecho efecto dosis-respuesta apropiado de la leptina sobre la señalización del receptor de leptina en el hipotálamo. Finalmente, se justifica investigación adicional sobre el uso de anticuerpos monoclonales como una herramienta terapéutica potencial para la obesidad.
Fuente bibliográfica
Antagonizing the Leptin Receptor in Obesity
Clifford J. Rosen, M.D.
Maine Health Institute of Research, Scarborough.
N Engl J Med 2023; 388:2291-2293